SPOT-peptide: Information
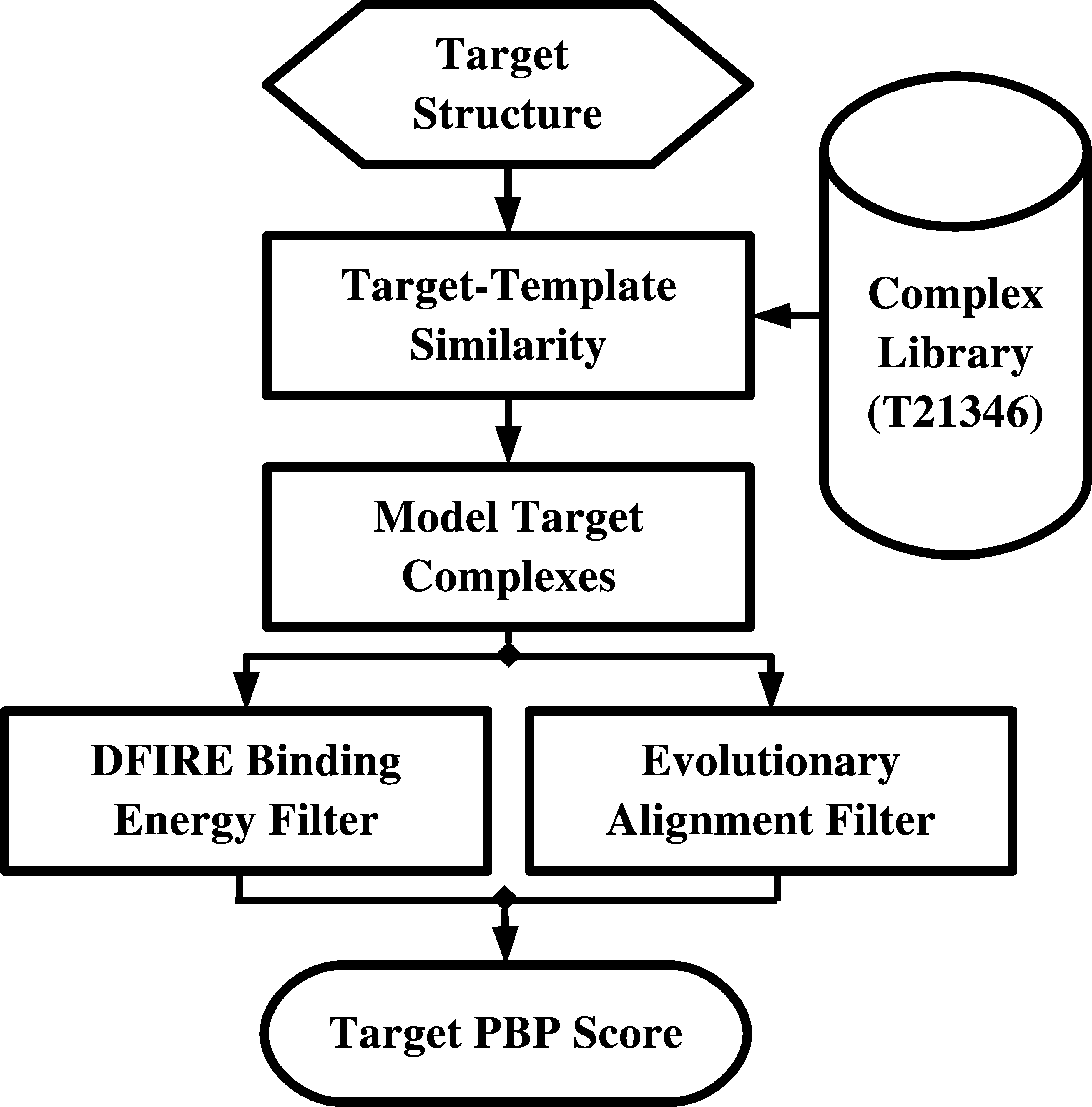
SPOT-peptide identifies peptide-binding proteins (PBPs) and peptide-binding sites based on structural similarity to known peptide-binding templates. The query structure is initially superimposed against all proteins in a library of PBPs. The quality of the superposition is evaluated by SP-score.
Model complex structures between the query protein and template peptide may be derived from top scoring superpositions. Potential PBPs are subsequently filtered according to 2 measures of local quality:
- A measure of binding site sequence similarity is evaluated by calculating the mean BLOSUM62 substitution score between aligned residues <4.5Å from the superimposed template peptide (EVO).
- The local consistency is also evaluated based on the DFIRE binding interface energy between the query structure and the superimposed template peptide (DFIRE).
Key terms:
Query: The name of the query structure.
Sites: The number of non-redundant predicted binding sites for the query protein (MCC<0.3).
SP-score: The measure of domain-level structural similarity between the query and top-scoring template.
TPL: The PDB chain ID of the top-scoring template protein.
TPL-peptide: The BioLiP ID of the peptide associated with the top-scoring template.
Cluster size: The number of redundant templates which imply the diven predicted binding site.
DFIRE: The interface energy between the query protein and superimposed template peptide normalised by peptide length (lower better).
EVO: Mean sequence similarity between query-template aligned residues in the putative binding region (higher better).
Residues: The predicted binding residues (based on original query PDB numbering).